
For a full list of BASHing data blog posts, see the index page. ![]()
Unwrap your fasta
DNA sequences are often stored as plain text in FASTA files. The formatting of a FASTA or fasta file is simple: each sequence is preceded by a description line beginning with the greater-than character (>) and containing information that identifies the sequence. A fasta file can contain many sequences, each with its own description line. Here's a 3-sequence example, sample.fasta:
>sequence1
gatgatacacgctgtcactaagcacattttctgtttctaatccataaccagaggcccggc
cccgctttttgcggatgcacaccatg-gataccttcgcggttctagcgatcctcgtac
gagaggtgctggtaaagtaacagtaatgggtactcccgaagtatactgttaccatcatac
accgccgtatttagcgaccgttacttgctcacctagatcctgtgcgacctcttggtcgat
ggtaatctctccactgcaagaagcccttattccgctgccacgactcatagcagaggtcat
acgtaacgaatctaaatccactactcacggctatggggtgaaatatt
>sequence2
gtcttgtatctagcccgctccggaacggcagctacgccacaggcaaacaatccatgcgat
gatcactt-cacaactggtgaatgatcgggctcaccaggacgtacagagaaagtggattg
acggagcatggtcgcaataagatatatacactggacccgatatgccctccattcctttta
cacaccctttactatggggtagttcggaggttgataatttgcggtc-ggtccaacggctt
cacg
>sequence3
gtgaaggtagttttgtgaatactgatgatcaagatgatatctcatcatcgcgttatgcgc
tgcaggctggttagcgtacgact-acaaaccagcacatgtacaagggtatgaccctctcc
aatgtggaacccttcggtagccccacaaag-
Applications that process fasta files accept either ".fasta" as the filename extension or a variation such as ".fna" or ".fas". On the command line, of course, no extension is necessary.
There are many specialised software packages for manipulating fasta files, not to mention online services that accepted uploaded or pasted-in files. There are also command-line fasta manipulators in the Linux repositories, in R and Python libraries and on GitHub. But since a fasta file is just plain text, you can do many of the common fasta manipulations with everyday text-processing tools like AWK and sed.
In this post I look at 3 AWK-ish ways to do one very useful manipulation, namely converting sequences spread over several lines into one-line sequences. In sample.fasta, above, the sequences are wrapped at a maximum of 60 characters per line. How to put each of the 3 sequences into a single line preceded by its definition line?
Method 1. I published this one by Bruce Horrocks in 2016 on the Linux Rain blog.
awk 'BEGIN {RS=">";FS="\n";OFS=""} NR>1 {print ">"$1; $1=""; print}' sample.fasta
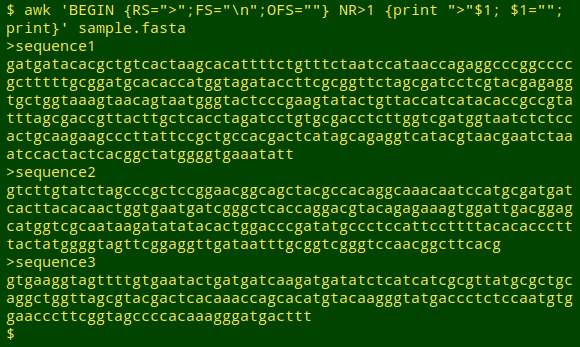
Paraphrasing myself from 2016:
The BEGIN statement sets the record separator RS to the '>' character. This means sample.fasta contains 4 records: [blank]>["sequence1" and its sequence]>["sequence2" and its sequence]>["sequence3" and its sequence]. Each of the 4 records is initially divided into fields (FS) with a newline, so that, for example, field 1 of record 1 is "sequence1" and field 2 is "gatgatacacgctgtcactaagcacattttctgtttctaatccataaccagaggcccggc".
Next after BEGIN comes a condition: the command that follows only applies to records after record number 1 (NR>1), so the blank first record (before the first ">") is ignored. The first instruction in the command is to print the character ">" followed immediately by the first field, so ">sequence1" gets printed.
The second instruction tells AWK that field 1 is redefined to be blank. Redefinition of a field causes AWK to redefine the whole record according to any earlier instructions. One of those earlier instructions (in BEGIN) is that the output field separator OFS is an empty string, and the sequence lines get concatenated by this simple redefinition of the record. This process is repeated for each record.
Method 2. A similar method was suggested by "melissamlwong" on the Cheatography site in 2015:
awk 'BEGIN {RS=">";FS="\n"} NR>1 {seq=""; for (i=2;i<=NF;i++) seq=seq$i; print ">"$1"\n"seq}' sample.fasta
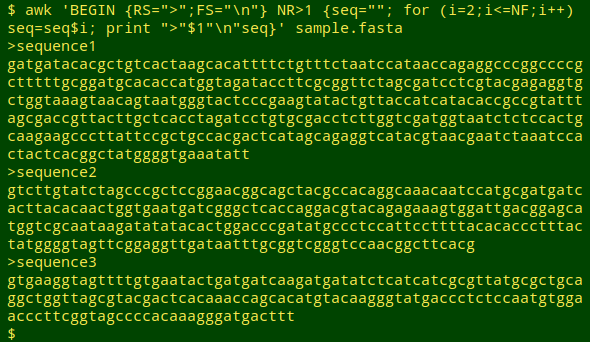
As in Method 1, the file is broken into 4 records by the ">" character, and the first (blank) record is ignored. In each subsequent record, the variable "seq" is first set to an empty string. Next, beginning with the second line (i=2), "seq" is redefined as its existing value followed by the whole line. This action is repeated for each subsequent sequence line. AWK then prints the ">" character, the first field (definition line without the leading ">"), a newline character, and the concatenated sequence stored in "seq".
In the original suggestion, there was an empty string between "seq" and "$i" in the redefinition of "seq", but this is not needed.
Method 3. The third method was suggested by contributor "Johnsyweb" on Stack Overflow in 2013:
awk '!/^>/ {printf "%s",$0; n="\n"} /^>/ {print n$0; n=""} END {printf "%s",n}' sample.fasta
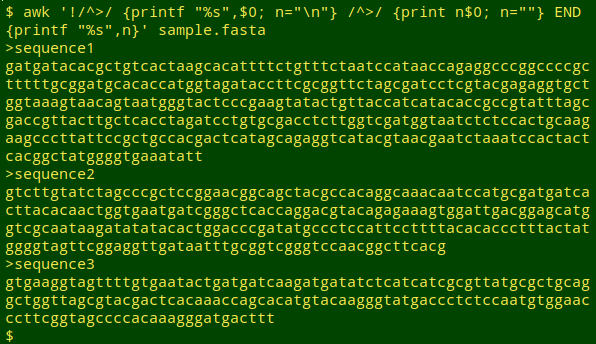
If a line in sample.fasta doesn't begin with ">" (!/^>/), then print the line without a line break at the end (this concatenates the sequence lines) and set the variable "n" to the newline character. If a line begins with ">", print "n" followed by the line, then set "n" to an empty string. After the last line, print "n". Since the last line in a fasta file will be a sequence line, this END command prints a newline character.
Caution! These methods won't work if the wrapped-line fasta file comes from a Windows environment and has a carriage return plus a UNIX linefeed for its line endings; i.e. "\r\n" instead of just "\n". Get rid of the carriage returns with tr -d '\r' < sample.fasta before piping the result to the AWK command.
Last update: 2020-01-13
The blog posts on this website are licensed under a
Creative Commons Attribution-NonCommercial 4.0 International License